
邻居老王最近有点麻烦。
老 王全 名王富贵,身患二型糖尿病,近期,老王携带的降压药所剩无几,于是,老王拿着药盒直奔药店而去,可是没过多久老王感觉血糖不降反升而且呼吸也变得急促了起来。
是不是买到了假药?
老王直奔市中心医院。医生拿起药盒对比了一下,告诉老王药品没有任何问题,只是两盒药的生产厂家并不相同,一个广州生产的,一个是河南。
为什么不同的厂商生产药效会有如此差别?老王听完,有点蒙圈。
其实,老王不知道的是,这就是中国制药行业的“潜规则”。
中国医药 行业的“潜规则”
所谓的“潜规则”那就是你当然不知道的规则。
在中国有很多如同老王一样的患者,对中国制药界的“潜规则”并不太了解——中国的化学药品有95%以上的药品都是仿制药。
仿制药,顾名思义,是一种在剂量、安全性和效力、质量、作用以及适应症上与原研药相同的一种仿制品。
通常,原研药的专利保护期过去之后,国家为了降低这些原研药的市场价格,各个国家都开始开放仿制药市场。理论上来说,这样开放的市场应该会促使原研药价格下跌,在欧美等其他国家确实如此。只是中国是个特例,原研药专利到期后,在市场上仍然保持高价并且销量不减反增。
这种仿制药市场增加了病患的医疗开支。
老王就是栽在了这个仿制药身上。
这到底是怎么回事?
原来,据GPLP研究发现,中国是除了美国之外的第二大医药消费市场,目前17万个药品批文号中,有接近11万的化学药品,其中仿制药占比高达95%。随着多个重磅药物专利的到期,中国仿制药正在迎接一个美好的未来,根据数据统计,2014年至2020年这七年里,将有约2600亿元的药品专利将到期,预期其中有46%的市场份额将被仿制药所代替。
而在全球制药市场上,自2013年起受专利悬崖的冲击,新药上市的速度大幅放缓,在此期间,全球各国纷纷出台政策来扶持仿制药市场,仿制药市场也在这段期间快速增长。值得注意的是新兴国家,仿制药在这些国家市场份额高达58%,远超发达国家16%的市场份额。
相比其他发达国家,中国无论是市场规模和发展潜力还是政策支持的力度都要远超其他国家,那么为什么原研药在专利保护期过后,价格降不下来,而且同类型的仿制药的药效跟原研药简直是天差地别?
原来这跟医药这个特殊市场有关。
仿制药行业的现状很大程度是由于十年前审批和监管政策宽松所致。
截至2018年6月13日,现存166043个国产药品文号,其中约95%的药品文号是于2007年前发放。值得注意的是,2002年至2006年是仿制药申报 高峰 期,主要原因是由于我国药品审评标准非常宽松——此话怎讲?
以2018年爆出的“神药”匹多莫德举例来讲。
匹多莫德,是儿科、耳鼻喉科以及皮肤科医生的“宠儿”。小孩发烧感冒咳嗽了,医生都会给开一盒匹多莫德。
数据显示,2016年匹多莫德在国内医院的销售额高达35亿元,在各大零售药店的销售额也达到4.27亿元,如果算上其他医疗机构匹多莫德2016年的市场销量超过40亿元,绝大多数的确由孩子买单。
就是这种给孩子吃的药在前不久爆出疗效不明确等问题。
原来,调查结果显示,匹多莫德最早于1993年在意大利首次上市,但是通过Pubmed和Cochrane知名的医学数据库发现,其参考文献不足100篇,而且集中于意大利、俄罗斯和中国等少数几个国家,而且排名靠前的文献还停留在动物研究阶段。但从国内数据库CNKI中搜索却找到超过1400条文献,其中许多还是核心期刊。
更为震惊的是,通过查阅FDA和EMA的官网,却并没有发现匹多莫德的注册信息,这说明尽管匹多莫德在意大利上市,却并未获得欧盟的认可,目前只在少数几个国家上市,中国就是其中之一。
“匹多莫德的有效性和安全性仍需进一步评估,在有可靠的研究证据出现之前,不推荐上市使用”,这是2016年该药准备在巴基斯坦上市时,专家组给出的意见。
然而就是这样一种药却在中国销量每年可以超过40亿元,并且儿童作为其主要的消费对象。
GPLP君对此嗤之以鼻。
究其根源,还是因为早期医药审批政策宽松所致的。在早期医药审批政策当中,曾有一段时间,为了推动更多的药品尽快上市,只要按照药品审评的要求,把各项材料报上去,很容易就批下来了,甚至有的药企造假数据过关。
也就是说,这些申报药品中有相当高占比的品种属于低水平重复申报,具体体现在申报企业的研究工作做的不充分,药厂直接从市场购买已经仿制的品种回来直接检测,用这些数据去申报,这对于当时很多企业来说则是千载难逢的机遇,企业只要能够按照要求做好申报材料就好了。
这与真正的监管实在差距明显。
比如,在美国,FDA对医药产品有一整套完整的认证 程序 以便确保新药的安全与有效.比如,首先,制药公司向FDA递交IND, FDA对新药的监测开始.此时新药的人体实验尚未开始, FDA主要审核体外安全数据与动物实验数据,以决定此药是否足够安全进入人体实验阶段;其次,药物要进行人体实验,人体实验共分4个阶段。一期主要测试药物的安全性, 主要副作用、代谢机理等, 样本数一般小于200;二期主要测试药物的有效性, 以决定药品是否能有效的作用于人体。同时,药品的安全性与毒副作用也是密切观察的对象. 二期实验的样本数一般小于300。
如果二期实验通过, 实验进入三期. 三期将包括不同的年龄段,不同的种群, 与不同的用药量, 以全面的研究药的安全性与有效性。三期实验的样本数在几百到几千不等.
四期主要在新药批准后进行, 主要测试药物的长期安全性, 新的种群等。
最后是制药公司完成了人体实验,验证了新药的安全有效性后, 正式向FDA提交NDA申请。 FDA审核全部的动物与人体实验数据,数据不合理,FDA会拒绝申理。
而在中国,中国仿制药的质量标准主要是看活性成分和外观、性状是否和原研药相符,而对给药途径、剂量、使用条件和临床效果上的一致性没有一个定量和定性的标准。
这让中国制药企业陷入“低研发投入,只能仿制国外药”的死循环。
不仅仅是药品,在药用辅料方面,辅料的生产质量问题也是导致仿制药质量差的主要原因之一。
在欧美,药用辅料的监管也采用和药品同样严苛的标准——据美国FDA的规定,仿制药生产企业在审批过程中必须申报药品中所使用的辅料,以及辅料生产企业的GMP和化验分析证明,并提供分析步骤。
作为药物制剂的基础材料和重要组成部分,药用辅料是保证药物制剂生产和发展的物质基础,在制剂剂型和生产中起着关键的作用。它不仅赋予药物一定剂型,还与提升药物的疗效、降低不良反应有很大的关系,其质量可靠性和多样性是保证剂型和制剂先进性的基础。
中国在2006年出台了GMP认证,但是当时的这个规范并非强制性的。可以说刚出台的GMP认证对这些上游辅料企业并没有什么约束力,这也使得当时的药用辅料企业良莠不齐,有很多化工和食品企业也参与其中,造成辅料质量的低下,直接影响了药品的质量和安全。
当然仿制药及药用辅料并不是根本原因。
如果严格监管,仿制药本身也不是问题,根本问题是“多级仿制”的仿制药。
这就相当于LV正品与高仿、低仿的问题,如果说创新药是LV正品的话,那么,高仿则相当于仿制药,当然这属于靠近正品的高仿,可以保证一定的质量,然而,如果多次模仿甚至低仿,比如说是“多级仿制”的仿制药,那么质量问题则无从说起。
也是,药效就会越来越差。
对此,为了保证药效,美国FDA规定,仿制药只能模仿原研药,相当于高仿。
然而中国则允许模仿已上市的国产仿制药,相当于“多级仿制”。
创新药的春天?
有差距的地方就是机遇,这意味着创新药的机会来了。
自2015年起,国家对药品审批和监管开始收紧,而且,2015年下半年CFDA启动的药品质量各环节的自查与检查到2016年3月实施的仿制药一致性评价都显示出,国家正在对仿制药实行高压监管。
2012年初,国务院提出仿制药与原研药质量和疗效一致的原则,随后也颁布了一系列关于仿制药一致性评价的文件和指南——2007年10月1日前批准上市的化学品必须重新进行一致性评价,而且必须在2018年底完成。逾期未通过者,药品生产批件将会注销。
据CFDA统计显示,2007年10月前批准的化学药品的仿制药口服固体制剂有289品种,涉及1817家国内生产企业。截至2017年5月,实际已经展开一致性评价的企业数仅占四分之一,放弃一致性评价的企业数量占比高达39%。
究其缘由,主要是 高昂 的一致性评价费用让这些中小制药企业望而退步。
可以想象随着仿制药一致性评价最后期限的来临,将会使得大量的药品文号退出市场,大约占总药品文号的90%。
其次,医保资源的抢夺将会成为未来仿制药行业的主要壁垒。
根据CFDA于2017年8月发布文件指出,鼓励临床机构优先采用通过一致性评价的品种,同品种药品通过一致性评价的生产企业达到3家以上的,在药品集中采购等方面不再选用未通过一致性评价的品种。
这意味着什么呢?
其实就是牌照的护城河。如此一来,通过一致性评价的药企,在招标定价、医保报销方面对比没通过的仿制药,就会有极大优势。
我们可以想象,一旦被纳入医保范围内,企业能否获益多少?
一方面,医保机构通过高品质仿制药来代替昂贵的进口原研药,可以缓解医保资金的压力,让有限的医保资金的使用变得更加高效。
这种双赢的局面势必会加速仿制药生产企业对医保资源的争夺。
当然,“两票制”的推行也进一步加剧了仿制药企业之间的竞争。
当然,“两票制”看起来只是减少医药流通环节,对医药流通企业影响最大,其实,对医药企业同样影响巨大。
国内的医药生产企业存在两种销售模式:一种是外企和品牌企业为代表的自营模式;另一种则是底价招商模式。
看起来“两票制”自营模式影响不大,但实际上公司的销售团队的结构和销售策略都需要进行大范围的调整,进而提升企业的运营成本。
另一方面,对于底价招商模式的生产企业来说,影响巨大,“两票制”实施之后,企业需要按照规定增加税务处理的成本和运营成本,使得企业最终只能通过提升药品的销售价格来摊销税务和运营成本的攀升,这也将导致一些以过票为生的流通商业逐步退出历史的舞台。
因此,两票制也间接推动了仿制药的淘汰及发展,推动了创新药的发展。
只是,创新药的发展并不容易。
首先,研发成本高昂。
据德勤最新报告显示,研发一个新药的平均成本已经从低于12亿美元增长至15.4亿美元,耗时也从10年增长至14年。
其次,失败率超过90%。
面对巨额投入,其高达90%的失败率也让中国的很多药企望而却步。
因此,在新药研发这项复杂艰难的系统工程,多年以来,中国的医药企业都扎堆在仿制药这个江湖当中。
最后,即便研发成功,如何让市场接受,这也是个问题,还不用说中国缺乏对创新药的保护,最终,面对仿制药这个江湖,创新药在中国依旧如履薄冰,如同蹒跚学步的婴儿,发展缓慢。
仿制药行业的江湖
有人的地方就有江湖。
仿制药领域也是如此,经过多年的发展,如今则是巨头 林立 。
资料显示,关于仿制药,其实门槛并不低。
首先,仿制药行业是个高风险、高技术含量、高附加值的行业,其研发能力就显得极为重要。他对比了三家龙头仿制药生产大佬们。可以看出这三家公司在研发支出占比都在上升,其中恒瑞在2017年研发投入比攀升至12.71%。
其次,医药企业的竞争逐步加剧。
数据显示,医药企业的销售费用占比目前正在提升当中。
以 恒瑞医药 为例,作为中国医药行业市值的“一哥”,早年是以仿制药起家,其最擅长的也是首仿药。早期的恒瑞医药主要是仿制广谱抗癌药和手术用药,如多西他赛、伊立替康、碘佛醇等。
截至2014年,恒瑞医药获批临床和生产的仿制药品种达到100多个,涵盖了肿瘤、造影、麻醉、糖尿病和心血管等多个医学领域,也让公司具备了丰富的产品线。前期积累的大量收益也为恒瑞进军新药研发提供了基础。2010年,恒瑞提出了“创新药+仿制药”双轮驱动的战略,并于2014年推出首个获批的创新药阿帕替尼上市,而另一个创新药艾瑞昔布也于2016年获批上市,现在恒瑞医药采取国际化创 新思路 ,让很多新药率先在美国开展临床试验,开拓海外市场。
另一位仿制药大佬—— 复星医药 ,在仿制药上布局同样深远。2017年底, 复星医药 在仿制药、研新药、生物类似药以及一致性评价等项目共计171项,2018年年初,苯磺酸氨氯地平片(5mg)通过仿制药一致性评价。根据资料显示,苯磺酸氨氯地平片(5mg)是其控股子公司 黄河 药业研发的化学药,该药2016年在中国销售额达22.6亿元。虽然仿制药的壁垒不是很高,由于生产工艺上需要规避原研药专利技术的一些产品和路线,也让研发难度激增,最终也是使得其研发投入巨大。
复星医药在发展战略上同样采取的是双轮驱动的“仿创结合”,其拥有的国家级企业技术中心,专门来打造具有高效的化学创新药、生物药、高价值仿制药和细胞免疫的四大支柱平台。
然而,即便这两家医药大佬,面对强势的医院,其话语权都相当有限。
当然,伴随着如今“医”“药”分家的现状,或许中国的医药企业话语权将进一步增强。
不过,如今,数字显示,这两家企业的销售费用占比都在提升。
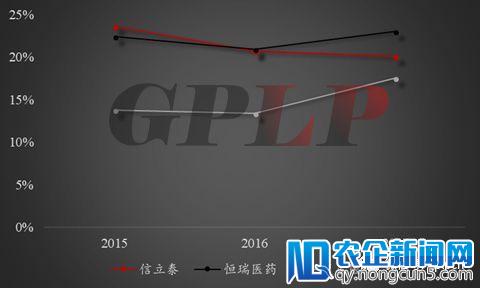
三家龙头仿制药生产企业销售费用占比情况

三家龙头仿制药生产企业应收账款占比情况(亿元)
当然,这也客观说明,除 信立泰 竞争力在增强之外,复星医药和恒瑞医药的竞争力都在下滑;应收账款占比则是只有恒瑞在2017年出现下降,这也说明其他两家企业对医院的话语权越来越弱,而恒瑞对医院的话语权却在上升。
更何况其他中小医药企业,因此,仿制药或者创新药市场虽然前景广阔,然而实现起来并不容易。
【本文为合作媒体授权 投资界 转载,文章版权归 原作者及原出处 所有。文章系作者个人观点,不代表投资界立场,转载请联系原作者及原出处获得授权。有任何疑问都请联系(editor@zero2ipo.com.cn)】